一文读懂日本医疗器械PMDA准入
医疗器械进入日本市场的注册登记流程较为复杂,且存在着语言方面的障碍,因此,日本一直被医疗器械制造商认为是极具挑战性的市场之一。为帮助国内医疗器械制造商顺利进入日本市场,思誉将为为国内医疗器械制造商提供产品设备出口日本的服务。
今天向大家介绍一下医疗器械PMDA日本市场准入的基本内容。
01法律法规
日本药品和医疗器械管理法律法规制度是《Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices》(简称:《药机法》)。
该法由原《药事法》变更而来。 法规由《药机法》、《药机法实施令》、《QMS省令》、公告、通知等构成。
02主管机关
日本药品和医疗器械的主管机关为日本厚生劳动省(MHLW)和行政法人医药品医疗器械综合机构(PMDA)。
03医疗器械的分类
1一般医疗器械(ClassⅠ)
由日本厚生劳动省大臣在听取“药事和食品卫生审议会”的意见后进行指定,这类医疗器械在出现副作用或功能损害时,对人的生命和健康影响的风险很小。一般指除高度管理医疗器械和管理医疗器械以外的医疗器械。
2管理医疗器械(ClassⅡ)
由日本厚生劳动省大臣在听取“药事和食品卫生审议会”的意见后进行指定,指除高度管理医疗器械以外的医疗器械。这类医疗器械在出现副作用或功能损害时,有可能影响人的生命和健康,需要进行合适的管理。
3高度管理的医疗器械(ClassⅢ、ClassⅣ)
这类医疗器械由日本厚生劳动省大臣在听取“药事和食品卫生审议会”的意见后指定。一般是指在出现副作用或功能受损的情况下(仅限于按照预定使用目的正确使用时)会对人的生命和健康产生严重影响,对这类设器械必须进行合适的管理。
04医疗器械的认证模式
1备案、认证和批准

2具体注册审核流程
所有打算将其设备出口到日本的外国制造商都必须在厚生劳动省 (MHLW) 进行注册备案。这种注册备案程序被称为外国制造商注册(FMR),以前被称为“外国制造商认证(FMA)”或“外国制造商认证(AFM)”。
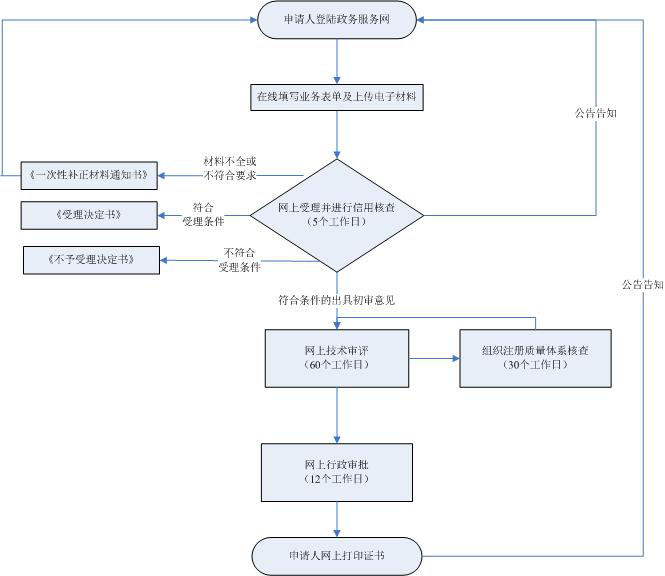
根据PMD Act& 第169号法令IS0 13485日本医疗器械注册审核流程如图所示:

05重点关注
1市场授权持有人(MAH)/市场营销授权持有人(D-MAH)
外国制造商必须指定日本国内市场授权持有人,这是作为在日本国内销售医疗器械的首要条件。MAH对日本市场上的医疗器械产品从批准、认证和备案,到上市后的安全管理负有最终责任,并对问题产品实施召回,且有义务管理和监督产品生产。除MAH外,PMDA允许外国制造商指定市场营销授权持有人,即D-MAH。D-MAH不拥有产品的注册和证书/批准的控制权,可视作为外国制造商驻日代表。
2STED摘要(注册资料)
STED摘要具体包括以下内容:
01产品规格
02稳定性和保质期数据
03性能测试数据
04风险分析
05临床数据
06制造(过程,监督,灭菌)
07遵守适用的标准和基本原则
08开发历史记录(先前的设备版本,全局授权)
3质量管理体系(QMS)审核
QMS审核是由医药品与医疗器械局(PMDA)或某一家注册过的认证机构(RCB)来执行的。QMS审核所需资料有质量手册、程序文件、ISO 13485证书、组织结构图、制造工程图、制造设备等。QMS审核范围包括制造销售商(MAH)、医疗器械的设计、生产制造相关的所有场所。是否需要进行QMS的实地调查,由认证机构判定是否有此必要性。
04多法规要求
外国制造商的医疗器械产品进入日本市场,还需要同时满足其他法规的要求,如电安法、电波法。


© 2023 深圳市思誉医疗器械技术有限公司 版权所有 粤ICP备2024264986号-1 粤ICP备16071228号-5